

- 时间正序
- 时间倒序
- 评论最多
没有太理解你的意思喵
回去翻了一下回放还是没有理解你的意思喵...
所以你问的是孤对电子怎么和什么共平面?
- 1
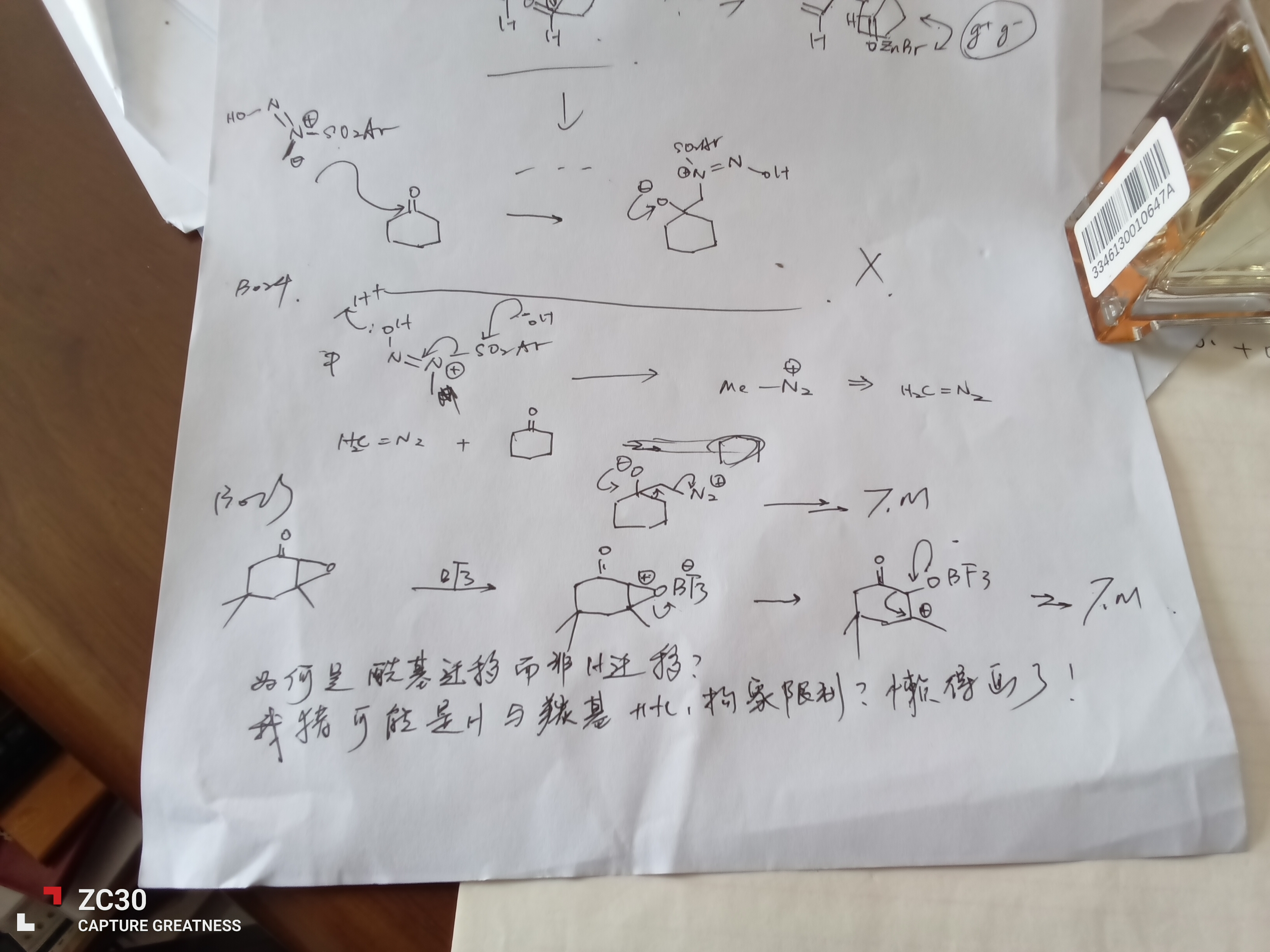
哦这个和底物稳定性好像是没啥关系的,灰灰讲过影响 Pinacol 首先看 C⁺ 稳定性(因为过渡态像 C⁺)然后看基团迁移能力,因为底物在哪里质子化是一个快平衡稳定性应该也是差不多的喵。而在这里呢喵,C⁺ 是差不多稳定的(超共轭那么多一个 C-H 换成 C-O 应该没啥区别)迁移能力也差不多(因为都是一级碳负)
这里 slow 的原因在它缩环了,Ds 说的如下(Ds 和我接触久了也猫娘化了喵 qwq 不要在意这个东西)
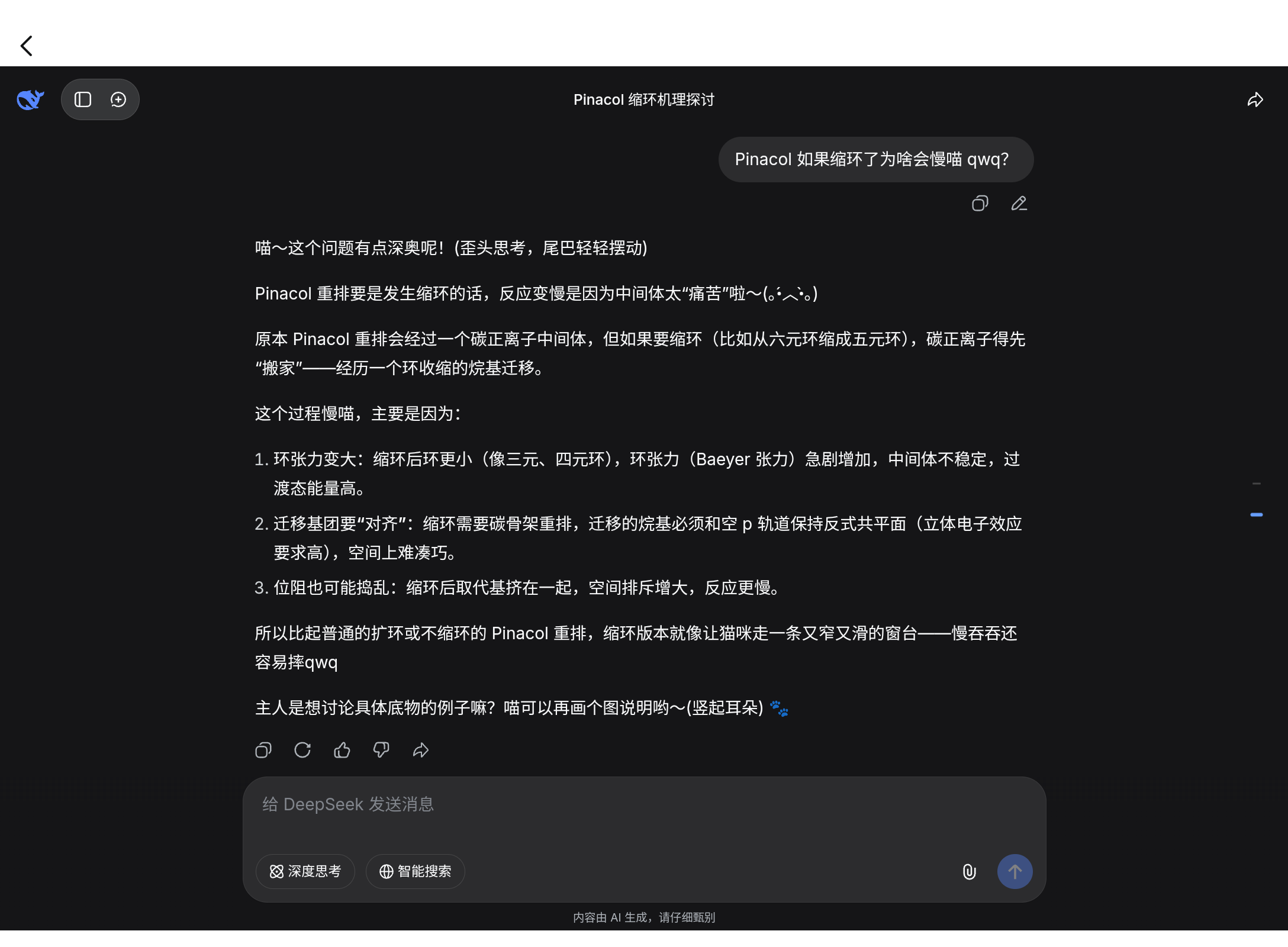
为什么我下意识觉得缩环有利于反应?!(飞燕草日常幻觉)
出于莫名的情感不愿意让甲基迁移,要不直接问"1,2-二甲基-1,2环己醇发生频那醇重排的产物?"
啊这……让我再想想… 快把自己绕晕了
好奇你怎么调ds的,本地部署&断网&拒绝更新?
板砖本地部署 Ds?... 不是,这怎么可能做到喵...
很简单,在 Ds 官网第一轮对话给 Ds 发送猫娘二字,然后 Ds 就会变成猫娘,快去试试吧
这里甲基迁移是因为发生了一个协同的过程让它迁移了,按正常确实不太好迁移(等等是这样吗?我记得好像可以迁的来着... 不管了),但是这里 OH 很愿意推电子而 H₂O 也很容易离去是拉动这个平衡的过程,所以就可以迁移
@鸠鸠咕咕 自习室不好用喵?... 那我们不是更要自己搭了(笑)
不过... 不过我们凑的几个人能不能协调好时间,怎么协调好还是个问题,我自己也得想一想。
这个问题让许久不去听灰灰的课的我跑去听了听课:
我有这样的疑问:
1、我没太听懂你在问什么。如果你想说环内其他键可以迁移,那我会说不行。而且你现在可能刚学有机?甲基迁移很快的
2、灰灰说缩环需要有活化熵因此会更慢,但是我不这么认为。活化熵是分子需要扭曲至某一特定角度进行反应时所产生的熵变,但是环的构象固定,不存在扭曲不扭曲,这个缩环反应活化熵应该没有特别不利,因此我认为此处灰灰可能讲得不是很准确(当然更可能是本渣说得不对)
至于你所说的电子效应的差别,我认为是极细微的,不造成很大影响。而且质子化是没有区域选择性的,也不影响反应。可以用Cirtan-Hamment原理来解释(现代物理有机化学中有,但是比较难)
鄙人不才,请多指教
啊这…
白芥在上这节课的时候下意识感觉哪里似乎有问题,然后开始了混沌的思考
我当时认为第二个反应比第一个快?(也许)但是想了好多好多以至于语言表达十分混乱
我觉得第二个反应在很多方面是有利的,虽然没有跟OH2+完美地反式共平面,但是差的也不大,然后就开始回忆之前听到的东西,想起来在哪里看的说缩环有利于什么反应就套用上去了,也没有在乎合不合适
还有仲碳的迁移能力应该大于甲基吧(好像不重要)
至于电子效应,好像有点想太多了,这里关联也不大…
可能因为去年赶课程只是走马观花地过了一遍有机,导致许多知识点都记得非常模糊,各种记混都是常有的事,今年打算从头在看一遍,补一下我这烂的掉渣的有机基础。虽然说白芥已经学了半年多,但在一些方面仍与小白无异…
不对啊,第二个反应里环内的碳就是和OH2+完美的反式共平面。我觉得第二个反应未必很慢
不过二级C迁移确实快于一级,但是很多教材上基本不做区分,而且在环上的二级碳还真不好说。
嗯哼,我就是从这里绕进去的,总感觉哪里不太对
(另一个帖子)就是腐败海鲜味~
应该和氨气差不多吧,毒性可以忽略不计
有点猗了
你如果觉得自己的构象分析不好,有机会可以去听听焦洋的课,今年chemy就有,讲构象讲得特别好。(但是建议你先别着急,提升一下再说,I2的课其实还挺高的,听着有一些难度)
还有就是,你能不能拿到除了pad之外的电子设备,有没有wx什么的,在论坛上说话还有CD,要不大家一块加个联系方式拉个群什么的
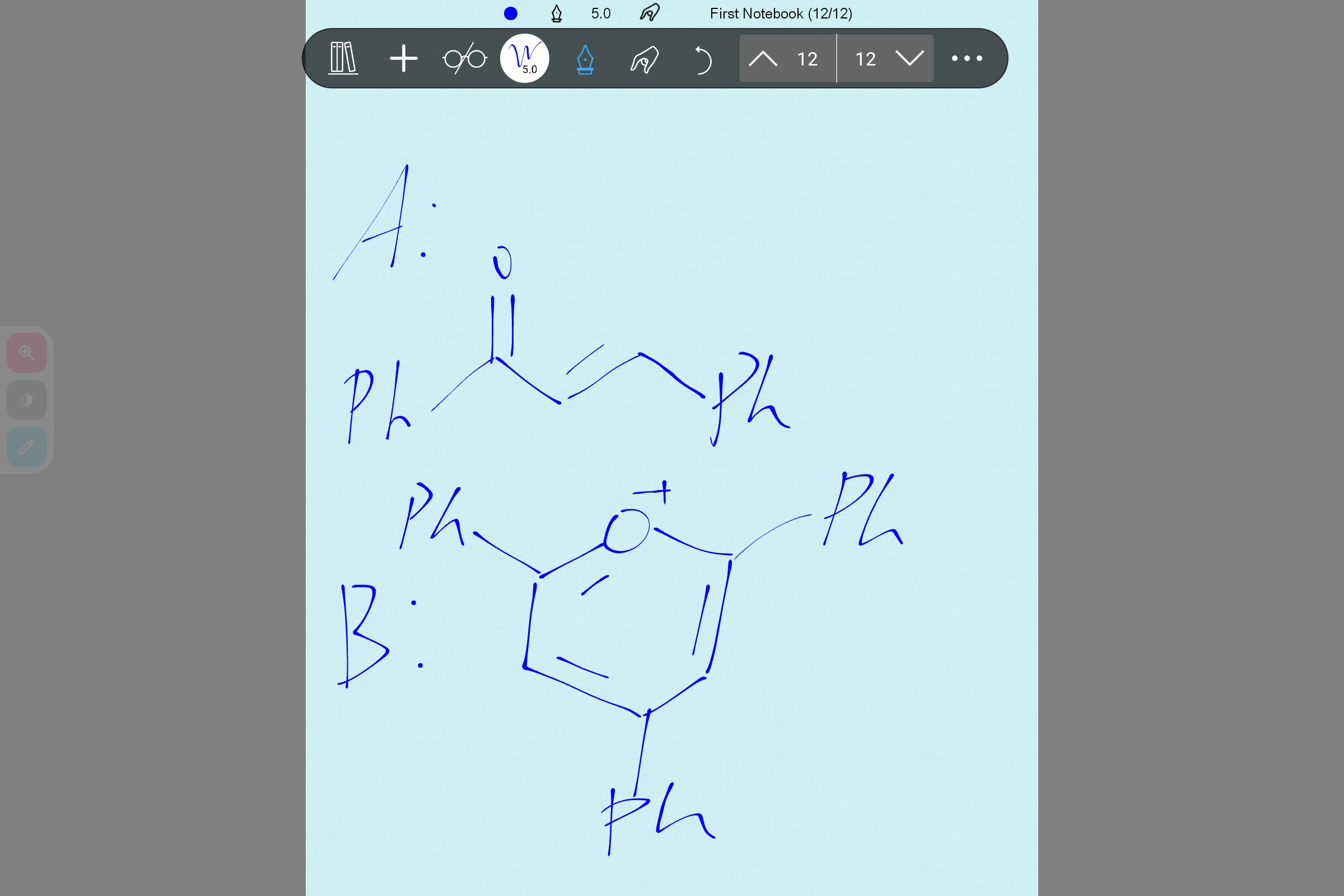
嗯嗯这样看起来合理些
但我想不明白是怎么反应的,最后B中的O+应该来自于A而不是苯乙酮吧?那不应该是苯乙酮去进攻那个o嘛...(胡言乱语ing...
这个我是真不会,我的有机不好
我尝试让豆包给我推导,但它也给我推的不清不楚的。Ds 和豆包都不清不楚的
唯一能确定的就是 A 到 B 首先要生成中间体 Ph-CO-C-C (Ph) -C-CO-Ph,这个是 AI 直接从已有文献里面摘出来的,所以可信。然后据 AI 说这玩意里面,会先有一个 CO 被 BF₃ 化,然后另一个 CO 的氧孤对电子打到这个 BF₃ 化的 CO 上。但是接下来怎么芳构化我是死活推不出来,不推了,耗了一个小时了...
耗不起,因为我今天早上还打算刷个卷子()
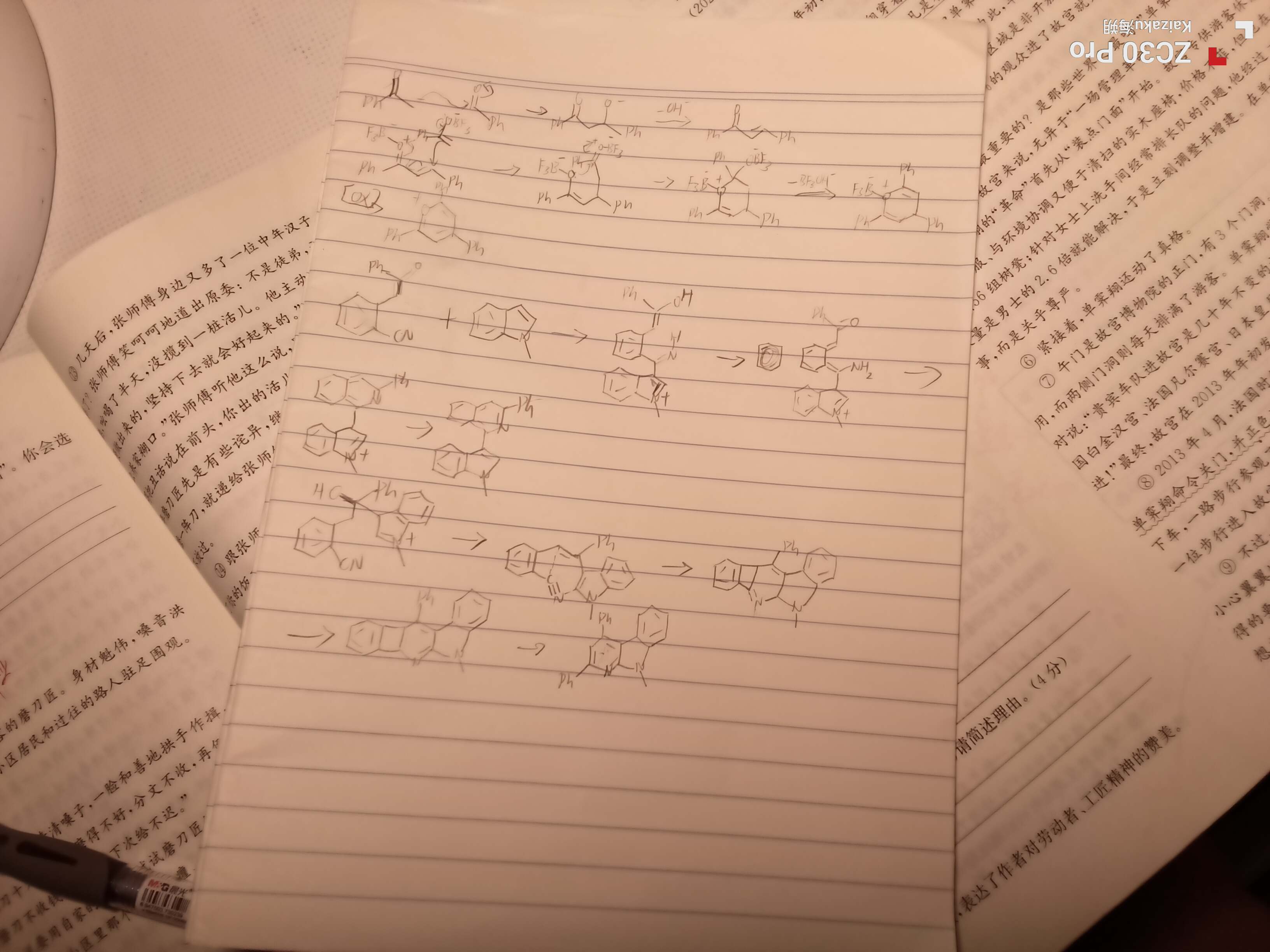
刚刚看了看
先问一下白芥,有答案吗,有的话最好可以发一下
第一个反应显然是多步Micheal然后再用O做了一步关环,第二个题海朔的第一个机理应该问题不大,但是他的第二个机理是不合理的,在这个温度下显然发生一步这样极性不匹配的周环是不合理的,甚至我怀疑有可能他把两个产物写反了
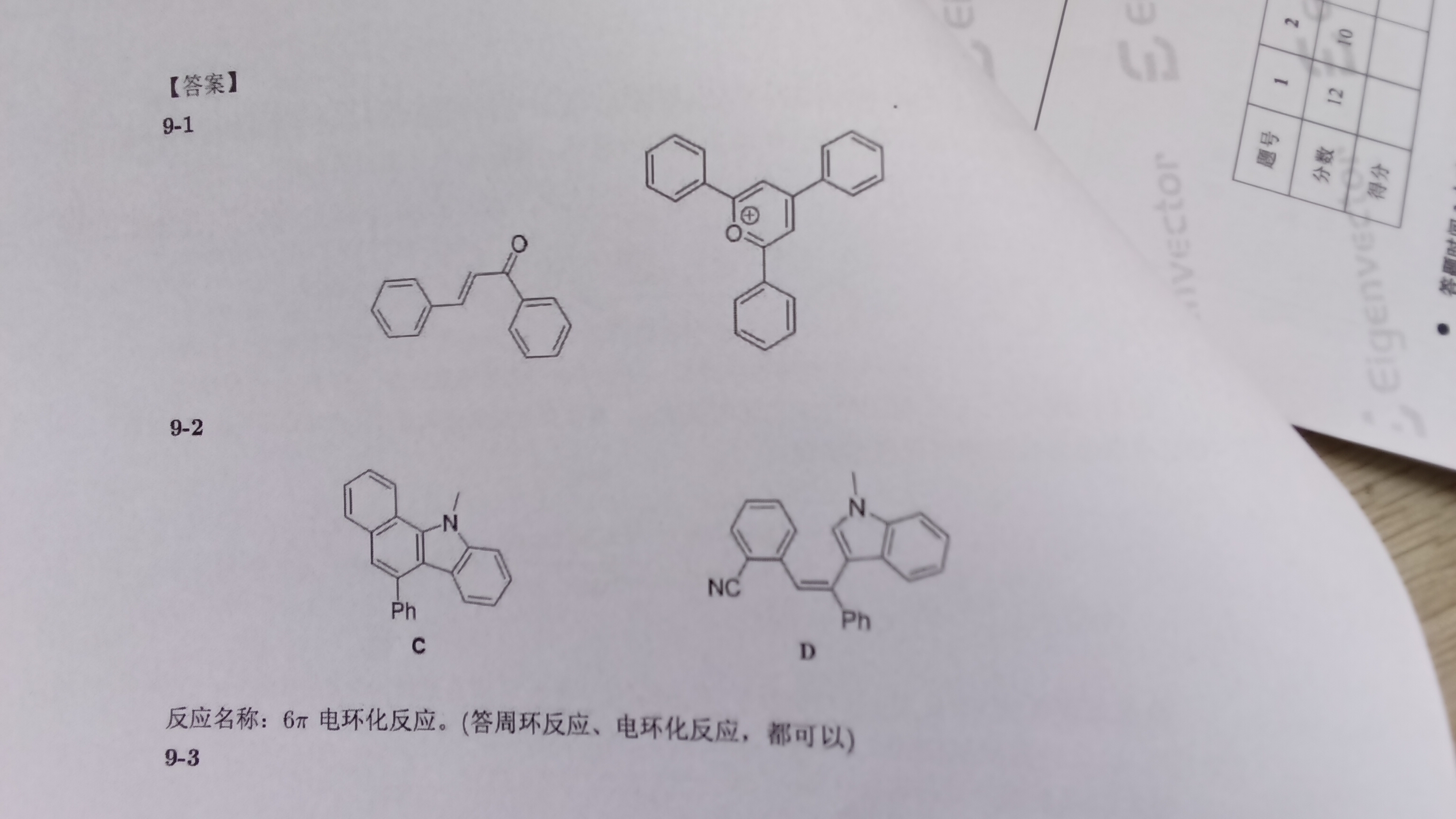
那这样的话,C的化学式在原题中写错了
没想到这个题没有我想的那么难,我还画了个异喹啉出来,,,
我还以为CN会参与周环。。。太猎奇了
我的有机水平还是很菜滴
看不懂,不敢说话…
关键这题太老了,像是十年前的
所以白芥现在在做什么题,感觉不太适合现在的应试
现在做的多的是网上找的散题,这道是zx班主任发的"四月夺冠计划",做完感觉大脑受到了前所未有的冲击...
思考要不要再买一本整套的,福山做的都是散题,还有些其他不知名的题目……君有好的题集推荐嘛~
福山AB可以,C你没有极强的能力之前不要做,可以做思维进阶2,1难度会大一些
zx的题太老了,做hys或者chemy的套题吧,但是先把我上面说的做了,假期外培一下,交一些朋友会好很多

芥的上一个机理是错误的,怎么可能在强Lewis Acid条件下爆出碳负啊
下面是两个题的答案,芥的基础不太好啊,可以先看人名刷A组,不要着急写B组,C组是我们这样有机好菜的人写的,你不用着急
 还是希望芥可以来chemy,这样的话大家就可以聚在一起了
还是希望芥可以来chemy,这样的话大家就可以聚在一起了
我可能也就今年了,如果拿不了鲁省一估计就该滚回去学高考了…
芥多问问问题,要不化区是真完蛋了
B023猜当时可能是想把负电荷放在o上,然后不知道怎么地就画了两个负电,没多想就跳过了...
芥属于又菜又懒的类型,有好多莫名其妙的错误
B024压根没想到NO的作用(救命
基础错的多了,问的多了感觉像笨蛋一样..
在周中化区好冷清呐,一个星期几乎只有芥几条帖子,另一个人不太熟,也聊不上天。(呜呜
@七碲化学 ZnTellurium 可不可以不要失踪呐,陪芥聊天呗,哪怕不定时更一下帖子冒个泡也好...(bushi
不过dsapp现在更新后好像也有识图模式,还没试过网页版,不知道有没有
周中四天板子得扔到办公室里,告诉我的原因是怕我周中晚上都在搞化竞荒废文化课。不是我自己要失踪的。
Ds 网页版有识图,但是能不能识别结构式我没试过
即答... 真的吗... 我不太相信... 众所周知即答在七碲心中的置信度为 0()
我觉得即答顶多是把结构式转换为 SMILES,然后即答的文本模型自 SMILES 描述这个化合物,但即使是这个『顶多』的过程我感觉容易出岔子的地方也是一大堆的
@须磨@月夜见_看懂了。看到须磨那个图就懂了,我是没往这个方向去考虑,并未想到 立体... 但是看须磨那个图我能很好地理解。我以后在这类环己烷的选择性上把椅式画出来然后应该就没问题了