

- 时间正序
- 时间倒序
- 评论最多
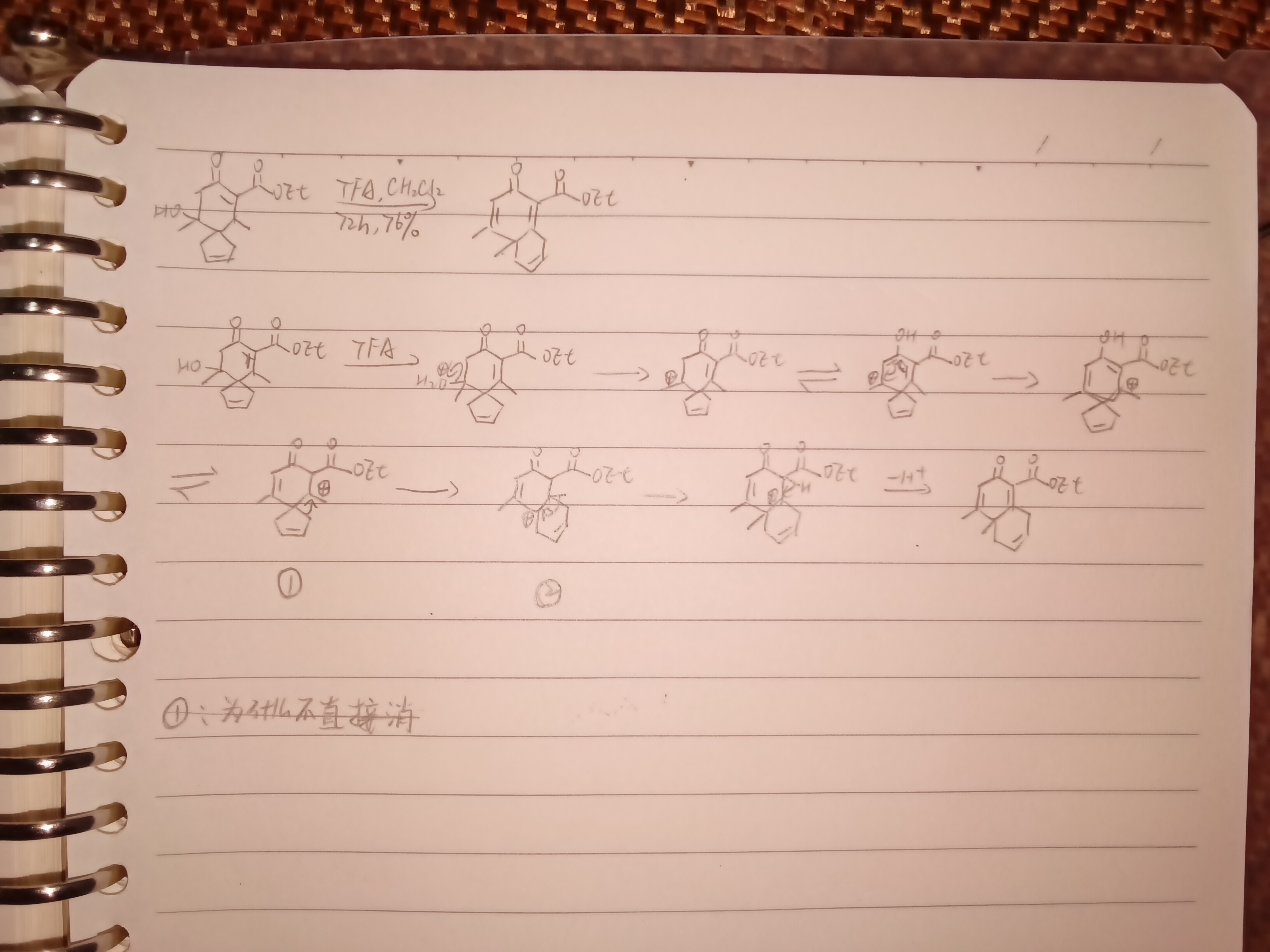
如图,①处为什么不直接消除羰基α位的H ,而要扩环,五元环不是也很稳定吗
②处为什么不消除下面烯丙位的H 直接形成大共轭体系
来源:有机人名(机理自己写的,不确定对不对)(还有就这么一个普通的碳正迁移还有专门的人名反应名称)
- 1
- 2
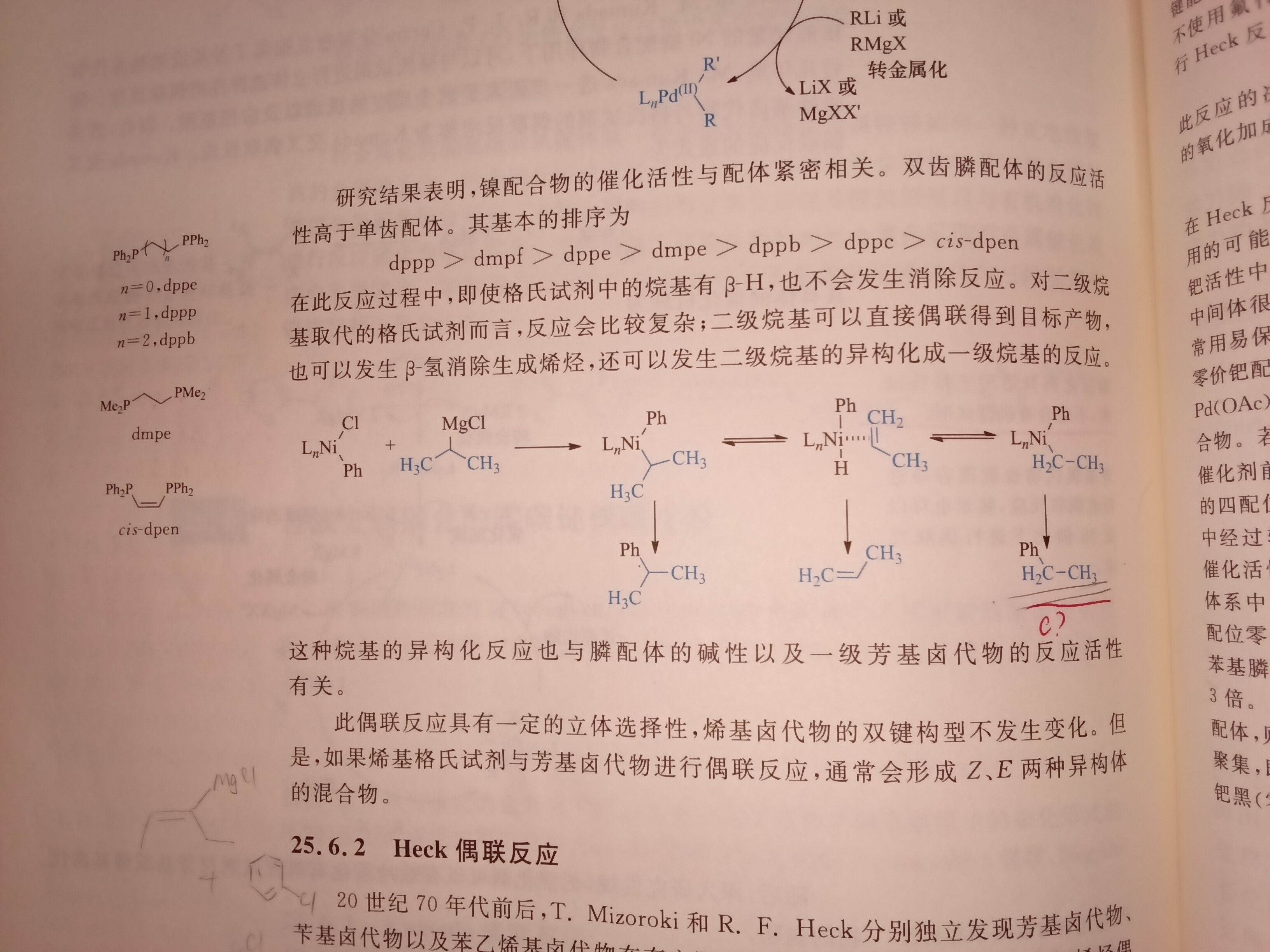
最后一步反应怎么把C 弄没了?

怎么影响的,求大佬画一张示意图
N 最后被氧化成什么了,大本箭头太抽象看不懂

高烯丙基和高炔丙基是什么玩意
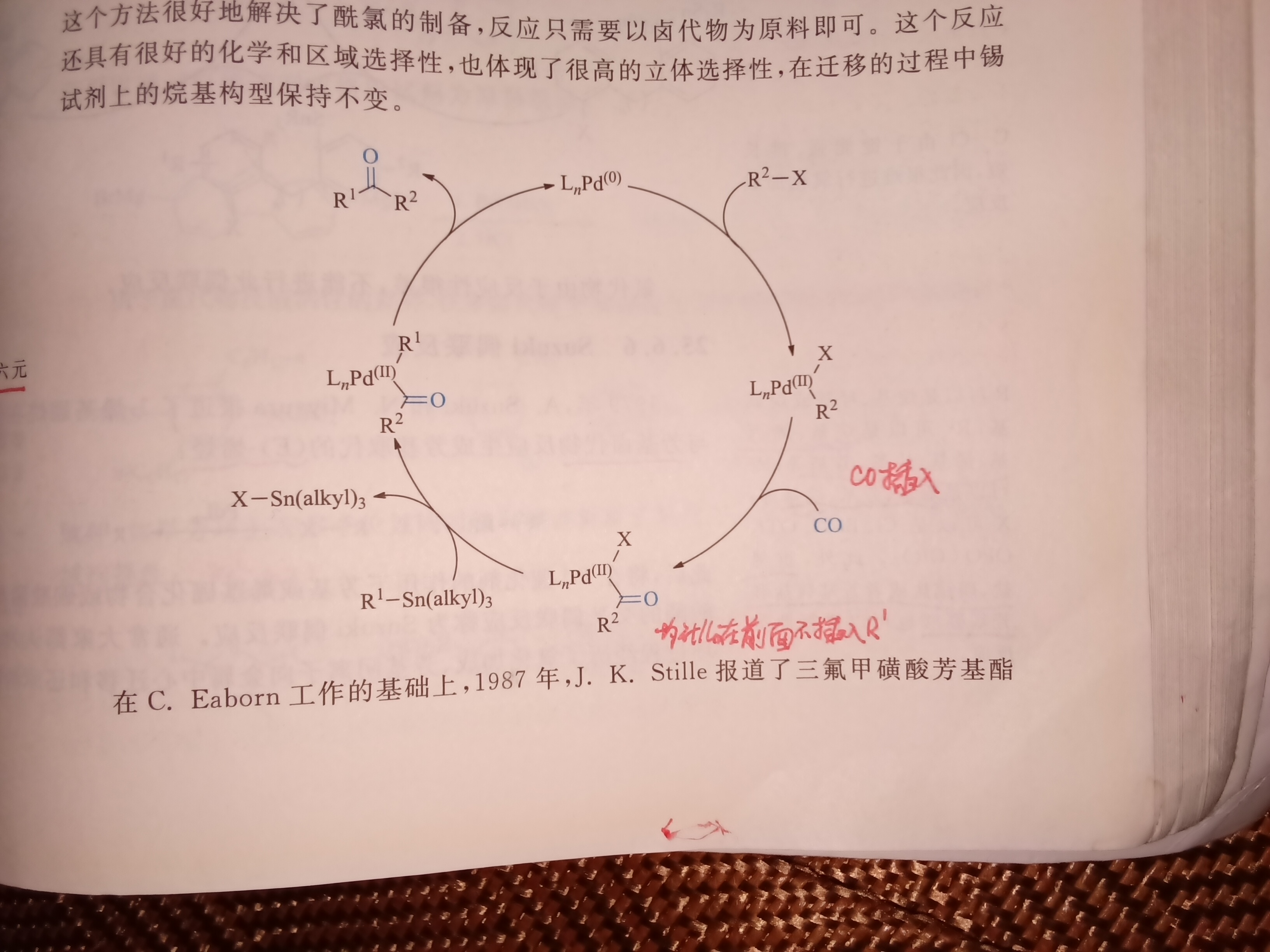
为什么不在上完R2-X 之后直接用R1-Sn 插入
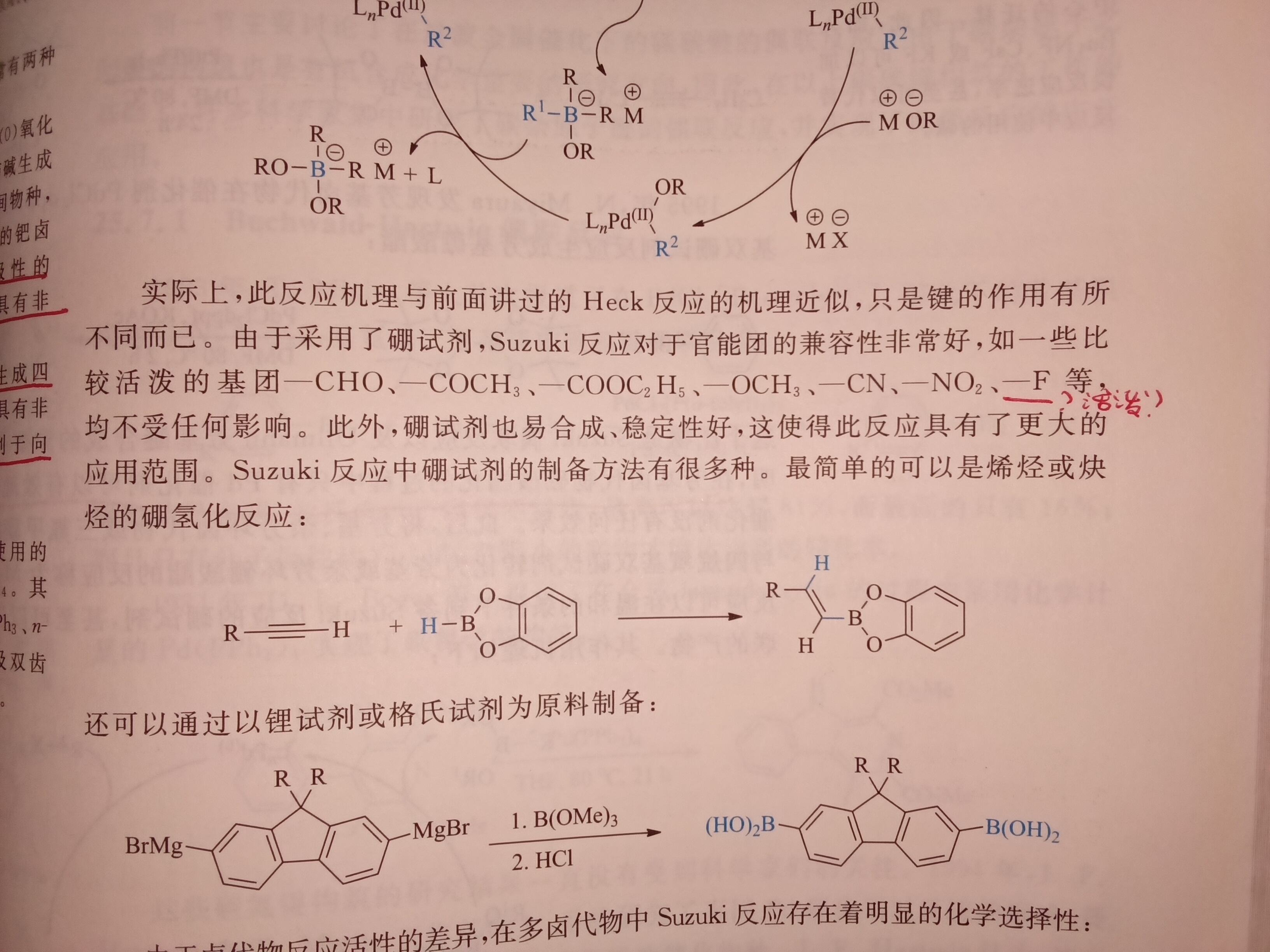
"-F是较活泼基团"…所以为什么?C-F 键能不是很大吗?
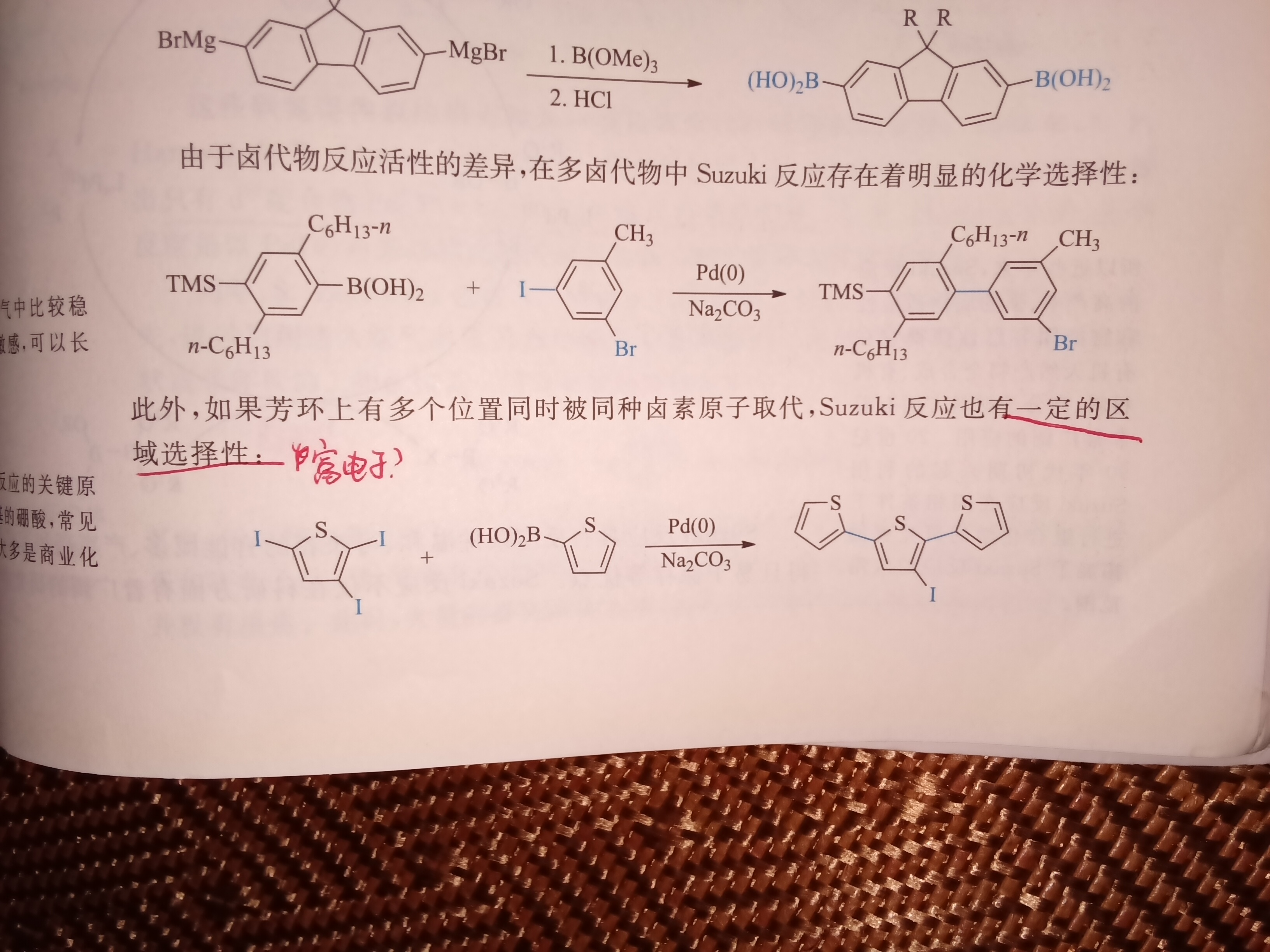
选择性来源是富电子吗?那原因是什么?不是说受电性影响很小吗?
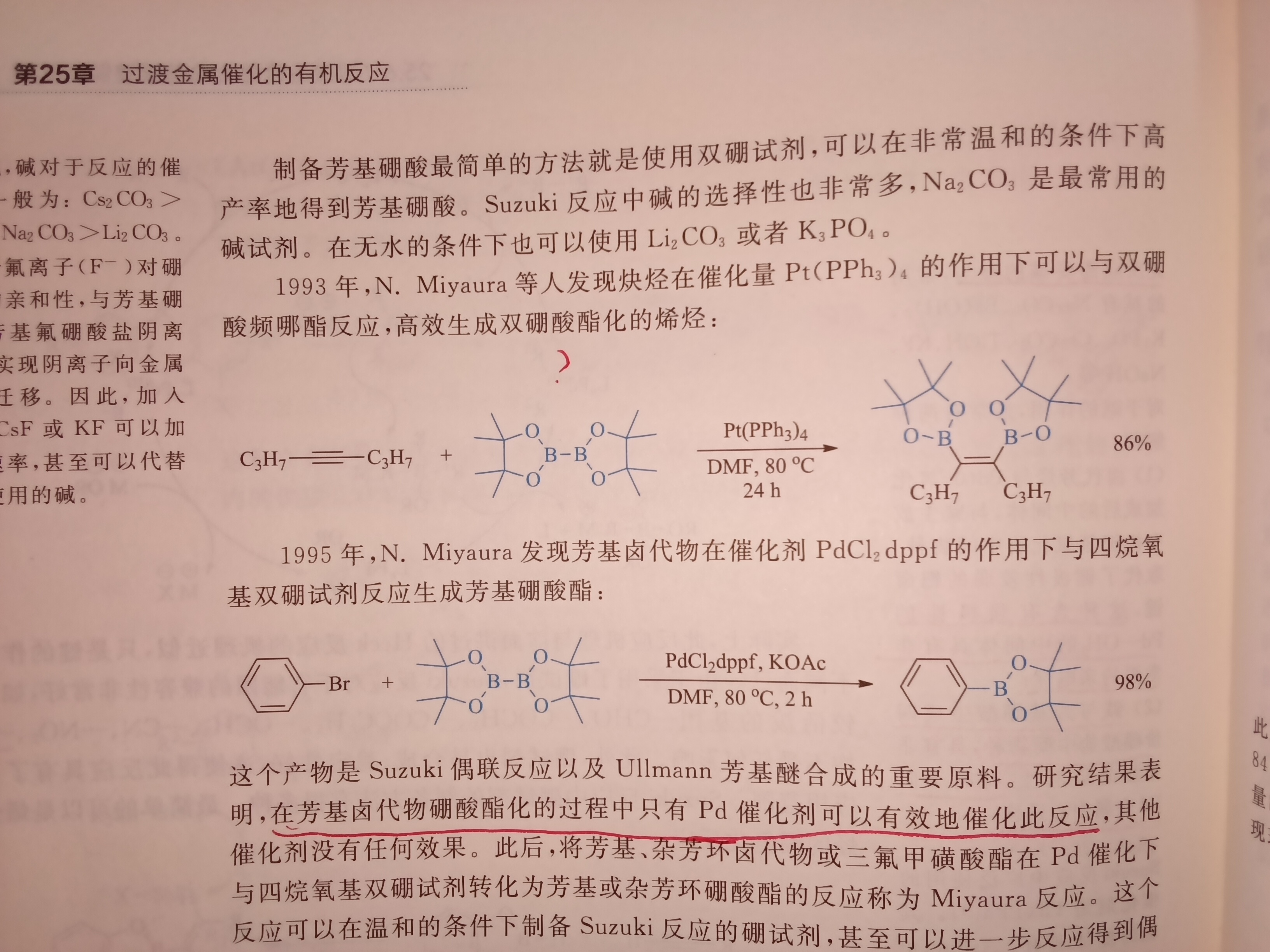
求第一个反应机理…

原因是二聚体不参与反应吗?那为什么前文说某人(懒得敲人名)认为二聚体是催化活性物种?
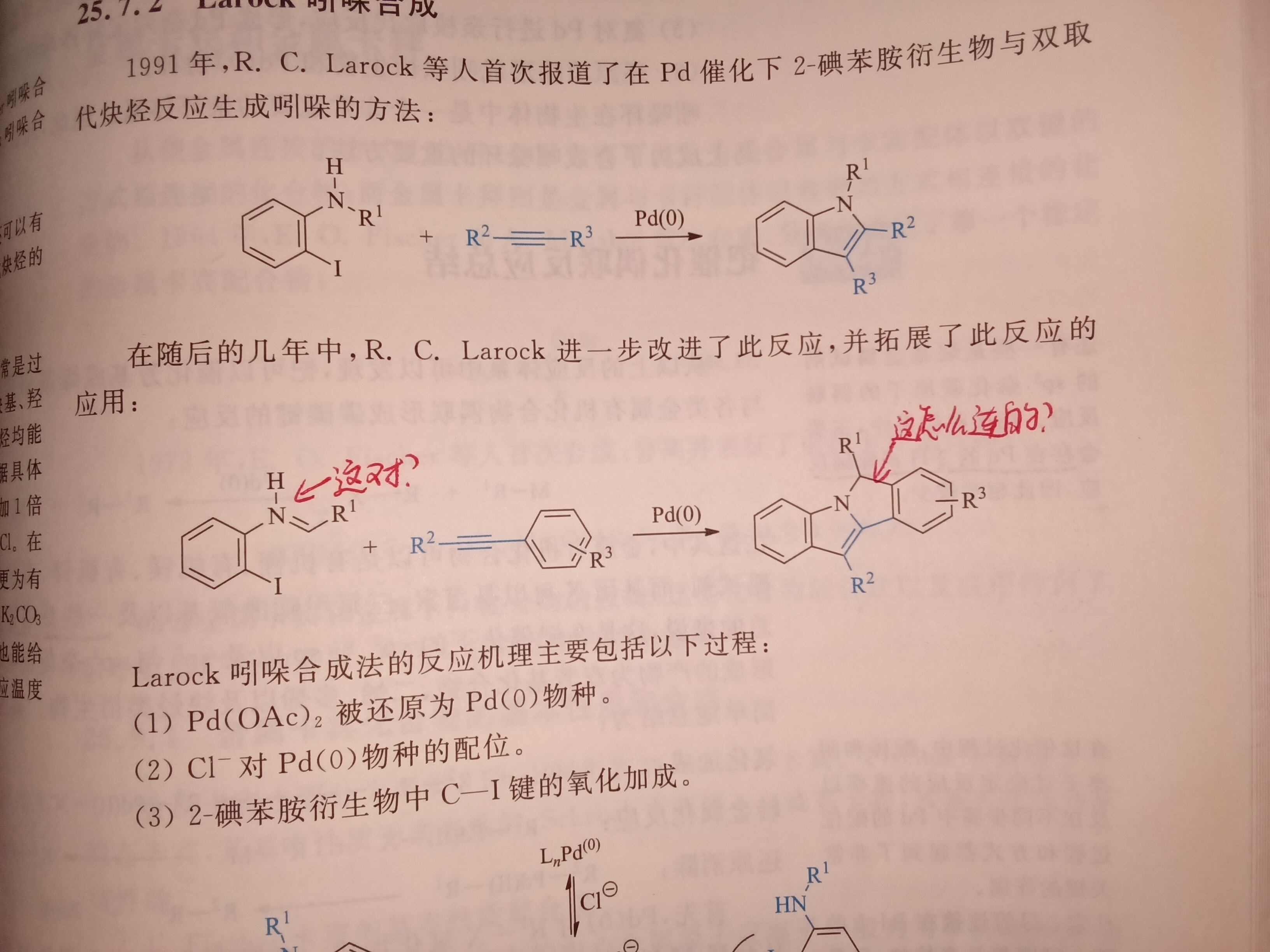
如图
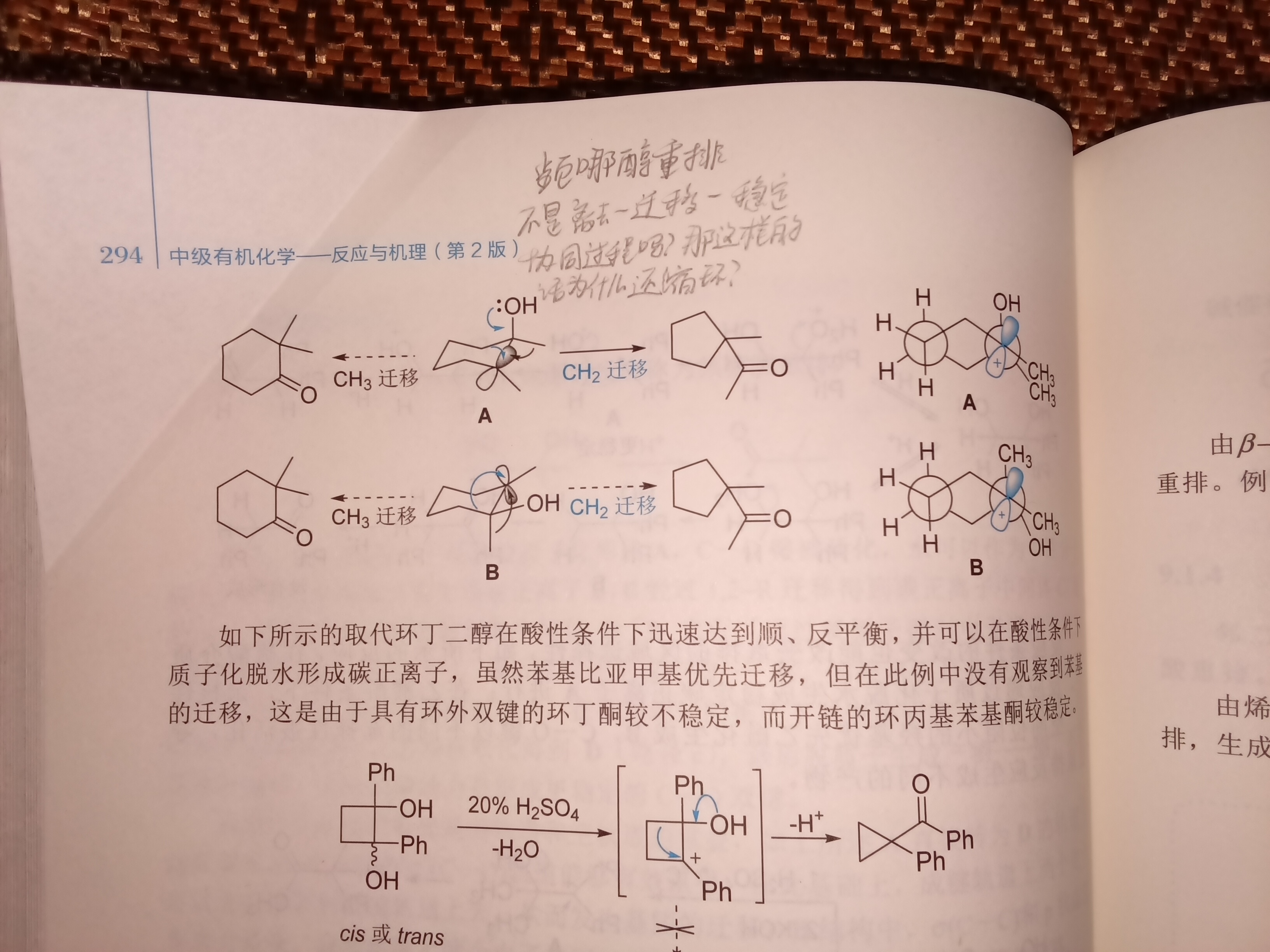
频哪醇重排不是离去—迁移—稳定协同进行的吗?那为什么还会产生C+离子并且甚至影响产物。大本上都写反式的这种醇是直接迁甲基的
还有,酸性酸碱怎么判断是软还是硬?
离子的变形性该怎么比较?
em....今天问题差不多就这样了(好像有点多…)
好多好多好多好多问题
我感觉我一个人回不完
氮那个最后氧化成氨基正离子,相当于掉一个负氢
等一下,那这样电荷就不守恒了吧。
(佬们是不是只有晚上才能上论坛啊?)
没问题
开始氮配体把一个醋酸根打下来了
哦,谢谢~
自习室还是挺有收获的,1,3,4,5,6,7,10都解决啦!剩下的等我晚自习研究研究(虽然明天期末然后我完全没复习)
谢谢各位大佬的帮助(不过今晚又会有一大堆问题出现了,嘻嘻😁)
@倚杖听江看到啦看到啦
em....同一侧是这个意思吗?(我不知道,只是问问)
个人觉得应该是类似顺式烯烃那种"同一侧"

内个,我竞赛一轮过了的💦
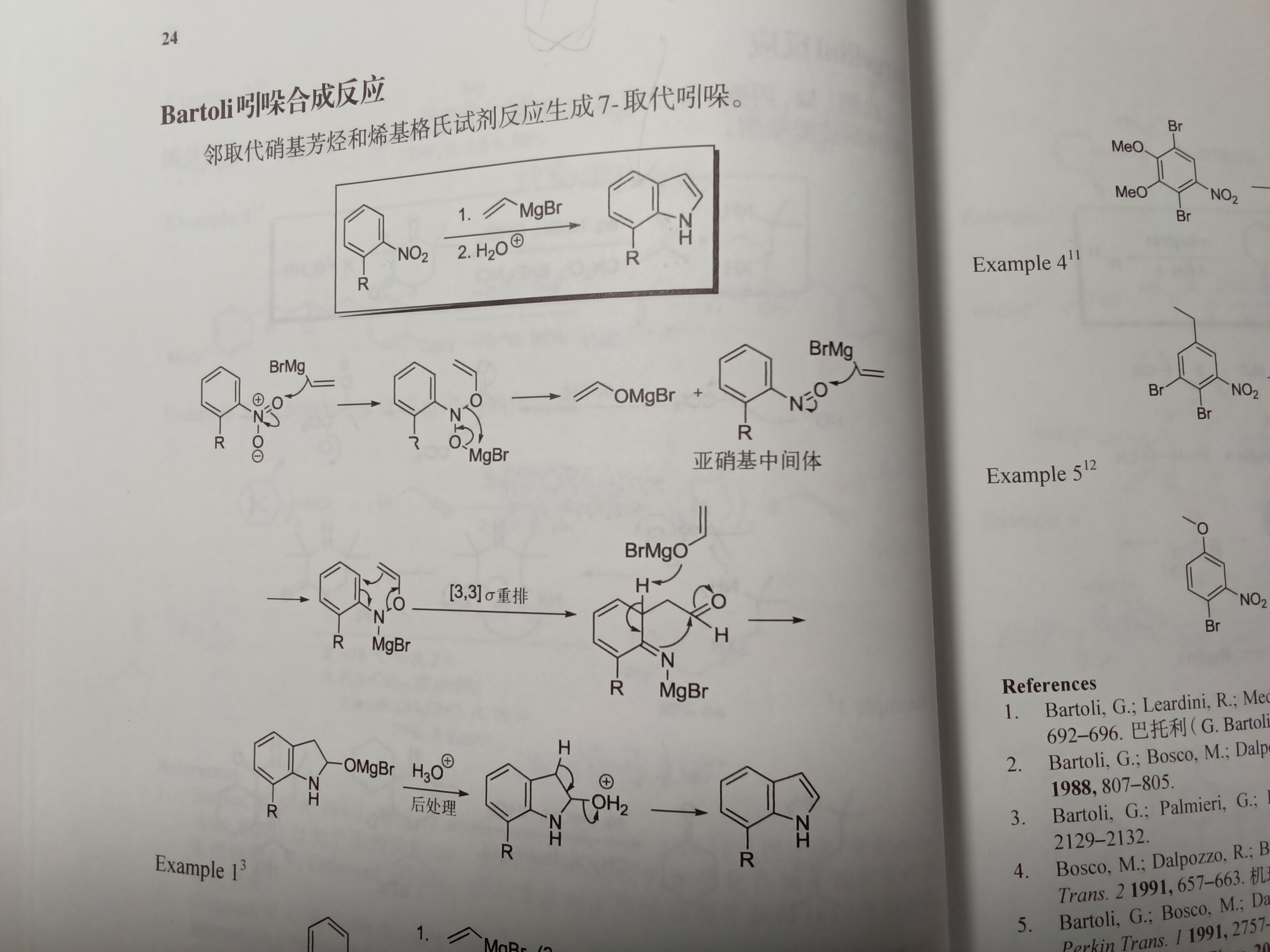
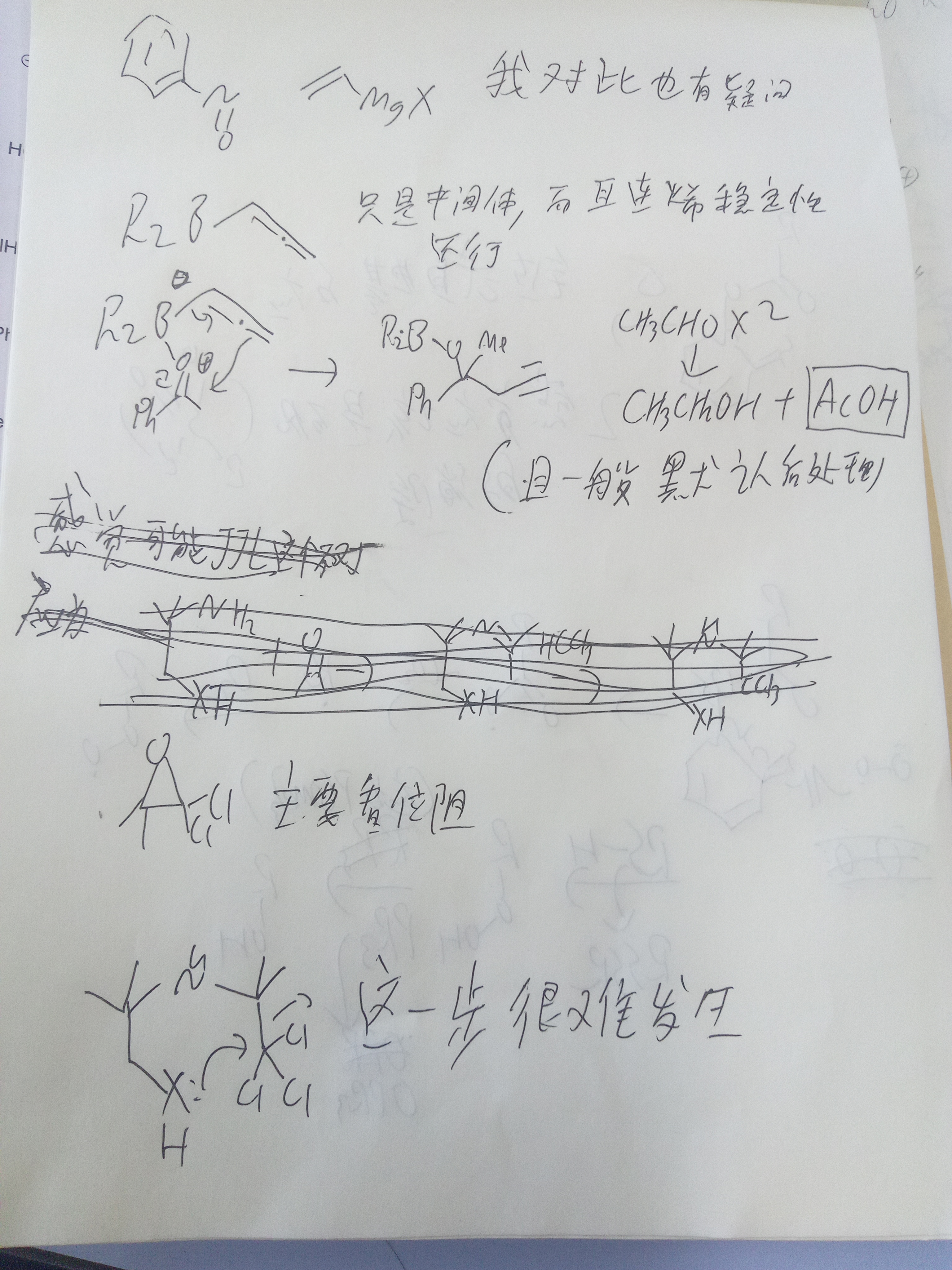
亚硝基那一步,为什么格氏试剂不进攻看起来更缺电子的N(和羰基一样)?
累计二烯不是不稳定吗(中间C电子云密度高)。然后整个反应我只能看懂它升温之前的步骤(不包括升温),之后是在干嘛?
为什么N开环的时候不开有Cl的那边?Cl不是有—C可以稳定过渡态里的"碳负"吗?
如图,上述反应为什么不能是这个机理,不会是因为Cl3C—亲核性更强吧

求机理。它都没有办法和硝基一样先做1,4加成再消除硝基
同类(?)反应机理如图
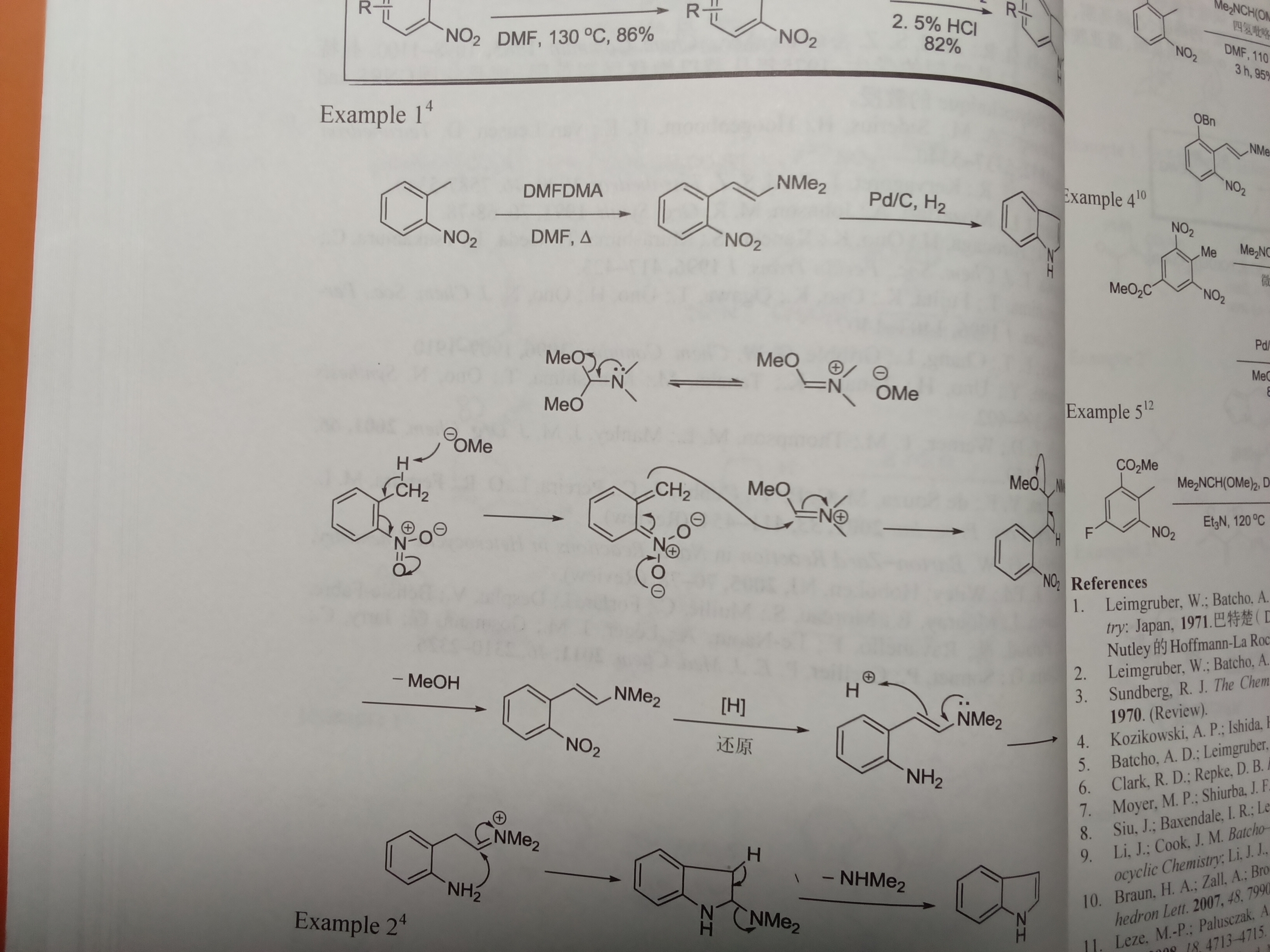
甲氧基负离子拔甲基上的氢?好歹一个是碳负一个是氧负啊
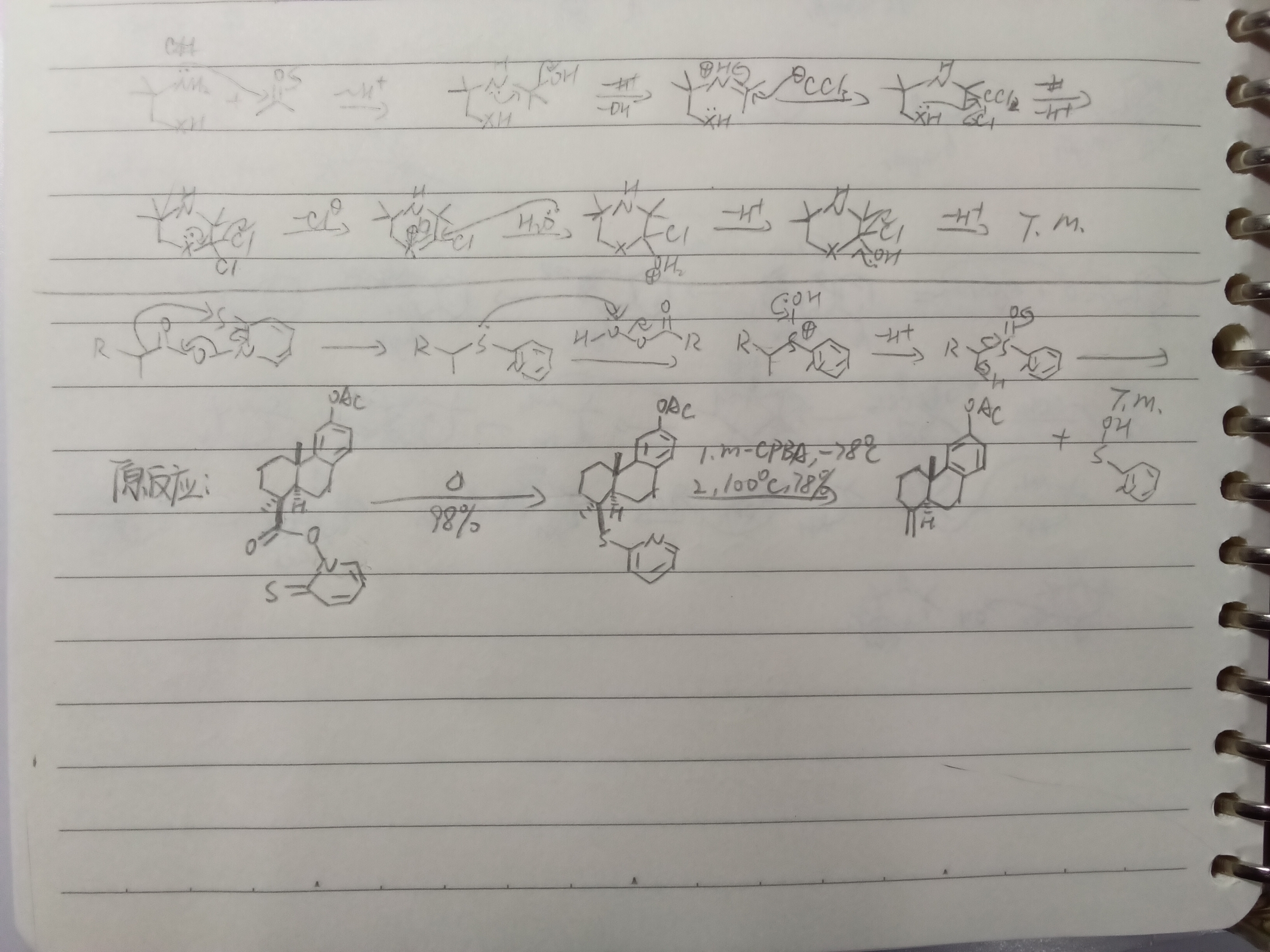
如图,这个机理对不对
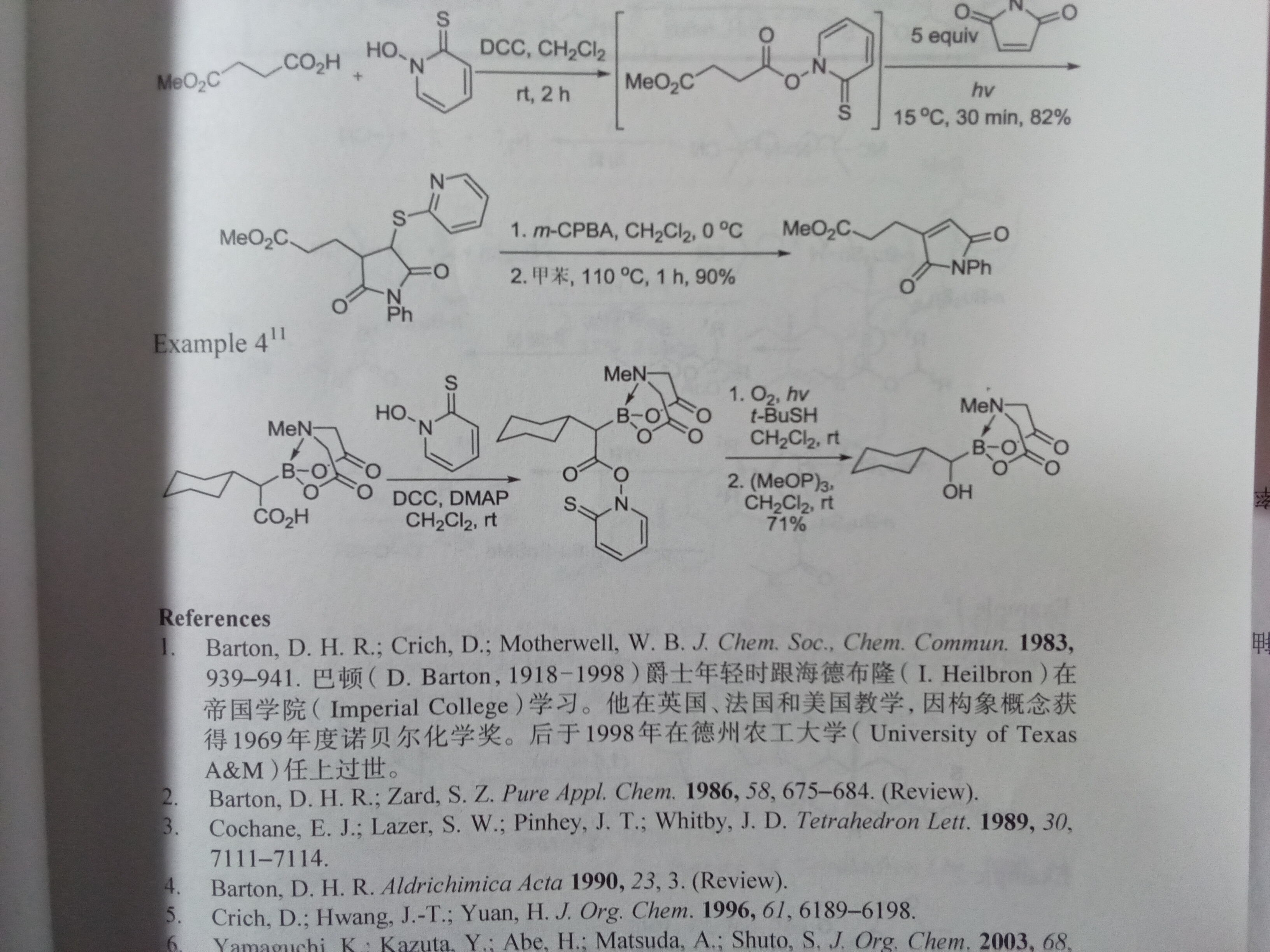
Example 4的后一步的机理,谢谢
今天就这些。
(为什么觉得写机理是最耗费时间的一项活动,总是写完几条复杂机理,一个小时就过去了)
其实我也就只是把问题放这,各位想回答就回答吧,毕竟两个月后都要国初了
楼盖太长了,下次能不能分开发,这样我容易看了上句忘下句♿♿♿
亚硝基是因为直接进攻N不能缓解其缺电子性,还是会有氮上形式正电。可以认为这里共振为氧宾氮孤对的形式,氧反而是缺电子的那个,进攻氧后N上不再具有形式正电荷
好的,那下次我一个楼就三题吧
等一下,亚硝基哪来的形式正电荷…(就是图上说的亚硝基中间体)
这两天考试,没时间看书…(啊!周末没复习导致考炸了啊!)
算了,复习我的英语去了(老师说很简单…希望吧…)
(之前的楼和这个楼要不要改一下啊?)
不好意思我看错了♿♿♿氮氧双键其实在哪一边断都合理,两边都有负电荷导致键不稳定,只能说看产物了
看人名建议看战略,裴sir推荐的,而且他非常反感JJL,就你那本(我也是这本橙色的)
原话是(你们可以把这个扔进垃圾桶里了
这…
那是哪里不好呢?太杂了?反正我就无聊看看,感觉和字典一样
PJ说这个机理有比较大的问题,而且例子不够详细,特例较多,也没有在合成中的应用
哦,谢谢!
那考试碰到人名反应应该是能通过正常分析得到产物之类的吧

qq你可以去b站搜化英社和chemy(chemy一般在小群讨论)简介有qq
N上有孤对,并不是亲电位点
画共振式出来氮正氧负贡献也并不大
可能是因为O是硬酸格式试剂是硬碱
我觉得可能并不能很简单的解释的通(自由基的话除非位阻非常大,一般都是氮连)
2.还是一样的,升温干什么用?
4.为什么难发生?N,O对于这么亲电的碳也不进攻吗?
第一步应该只是配上,升温才33,被多个卤素取代的碳是惰性的,极难发生除成3元环以外的SN2
哦。不过为什么惰性?
位阻。
哦,懂了,谢谢!
《真的好多了》
???
你不是评价他写的机理很合理吗?
啥机理,打氧那个吗
关于Bartoli吲哚合成法格氏试剂进攻位点的讨论结果:(以下来自化英社大佬4月的讨论)(自己总结的)
本来氮比氧略微容易被进攻一点,但由于氮旁边有位阻,格氏试剂只能打氧。但好像其他碳负不一样?
以下是原始聊天记录:
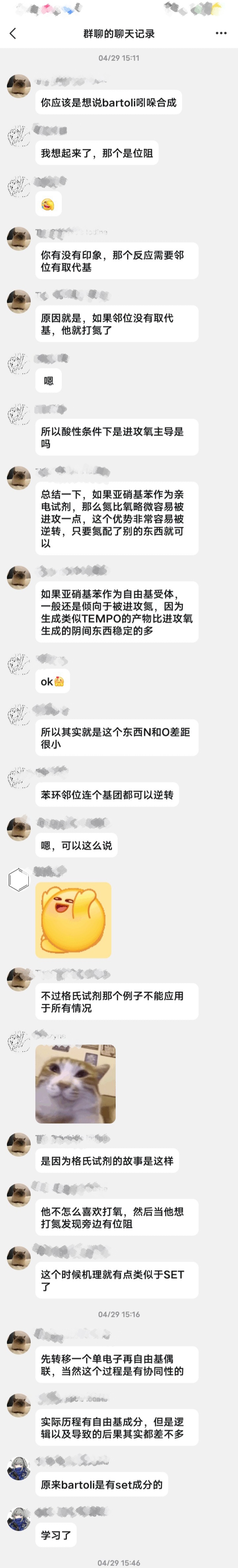
当然还是有问题的,要不然为什么叫问题帖(bushi):
1.为什么亚硝基苯氮和氧被亲核的优势是差不多的?是不是苯环共轭缓解氮缺电子?
2.进攻位点的选择性不是由氮位阻决定的吗,那为什么不适用于其他情况?是不是碳负位阻也有较大影响?或者大佬在这里只是为了强调机理的差异?
啊,大佬还是太超标了。他似乎认为我看得懂全英文的文献…(没有喷的意思,就是本人英语太菜了😭……)
其实不用打码,都认识()我印象中好像还提过Mg先配到N上的机理
不太对吧,按道理如果苯环共轭,那N和O电子云密度应该差不多,那Mg配哪就没有太大的选择性吧(除非又软硬酸碱)
但是这个反应确实产率不高40%-60%
em....确实可能有这个机理。大佬刚说了有LA的时候都进攻氧。发言冒失了…
@跳跳康 @跳跳康(为什么有两个?) @倚杖听江[千蚊斩] @Sbchem 请问一下chemy的卷子怎么找啊?
em.....还有《战略》全名是什么....
上mingke.xyz
@跳跳康我只看出来那个猫猫头像的是碘神
问的人是青岛二中的



所以具体就是由自由基机理对位阻的高度敏感,导致有位阻的时候只能加成O。
但我还是有不清楚的地方(有的是单纯学识不足…)
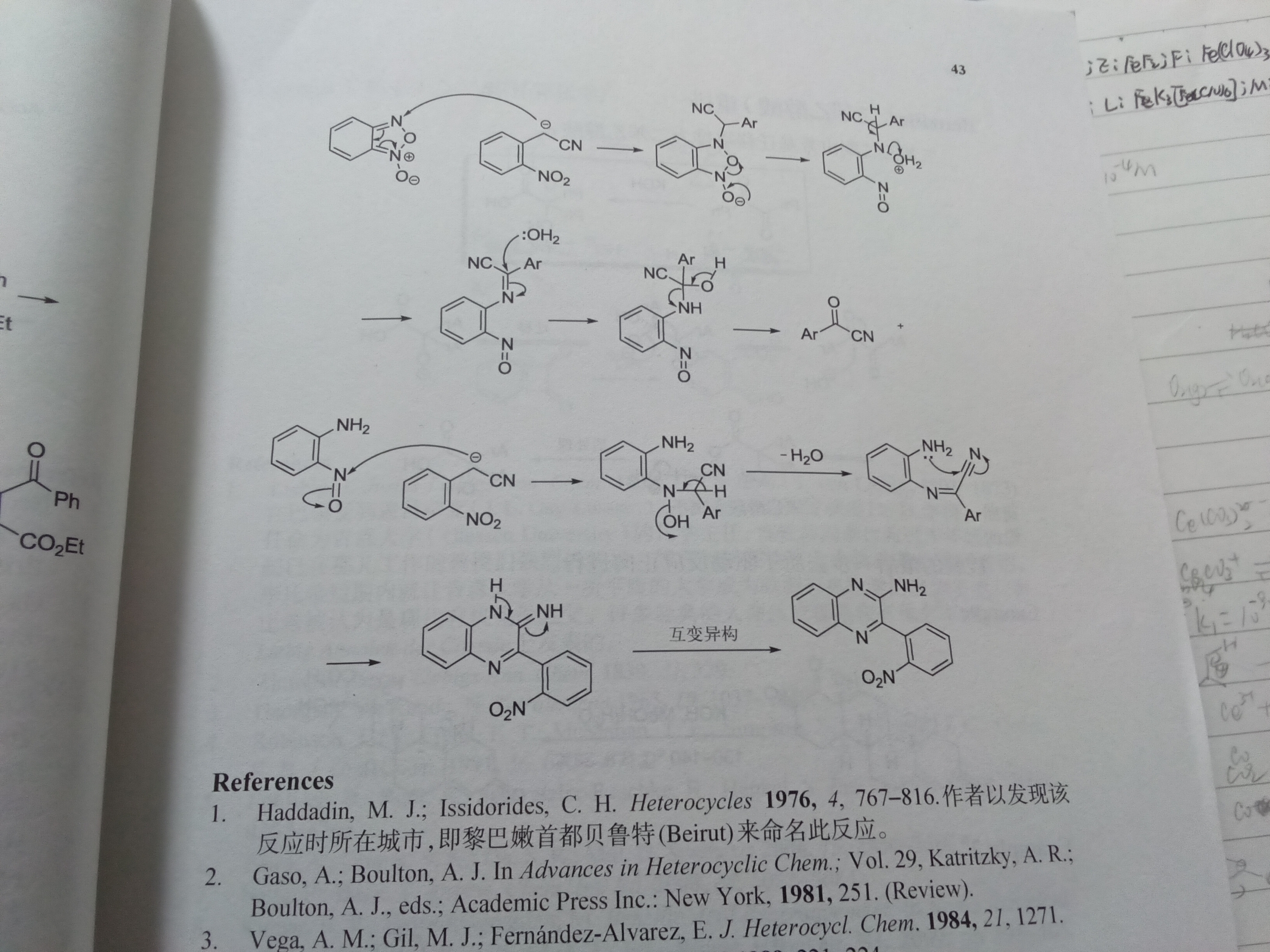
1.几个名词:苯胲,CH规则
2.亚硝基里N对O的—C哪来的?
ch原理看灰灰的250,我不太能打字说明白,苯胲刚刚用ai查了下,是苯基重氮阳离子
π34变为π22之后(π键的)homo和lomo都降了,更容易发生单电子转移
或者说取代苯甲酸里,如果有邻位效应,酸性就一定最强,不考虑氢键之类的
羧基两边都有取代的酸性为什么比邻位单取代的酸强?
单取代不是已经把羧基顶上去了吗,再加一个之后位阻影响不是和单取代的一样吗
最近又没人了啊喵…
为什么这里不可以直接用草酸滴定过量高锰酸钾
来源:GChO7,T6。大致为用KMnO4测COD。
我突然发现CrO3氧化烯丙位成α,β不饱和酮的条件可酸(CrO3(1.7eq)+AcOH(14eq))可碱(CrO3(20eq)+吡啶(140eq))。
2—碘吡啶为什么不能做亲核试剂啊…